Website
The main interface is a one-page application. You can upload fasta files in the upload aera.
The clermontTyping method is described in Beghain J, Bridier-Nahmias A, Le Nagard H, E.Denamur and O.Clermont. Clermontyping: an easy-to-use and accurate in silico method for Escherichia genus strain phylotyping. Microb Genom. 2018;4:1–8.
For getting help about the output go to the Interpret output section.
What is the format of the input files?
Fasta. The files have to be at fasta format, one file by sample, and the sequences can use IUPAC code.Is it possible to analyze multiple files in one time?
Yes. Up to 10 files are allowed in the same time. You should have only one genome by fasta file.Take care to name your files correctly! If several uploaded files have the same name you won't be able to distinguish between them in the output!
Can I upload raw reads from sequencer (fastq file)?
No. The clermonTyping method only works on pre-assembled contigs. For assembly you can check for others programs suchs as SPAdes or ngopt for illumina data.Is there any limit in upload?
Yes. The maximum size for a file is currently 10Mb. An E.coli assembly should be under 5Mb. If your file is greater than 10Mb, this is most likely a poor quality assembly. In this case, try to manually remove smalls contigs to reduce file size.Advanced parameters The best parameters for phylogroup typing are already set. If you want to customize your analysis consider trying the standalone version.
Nevertheless, you can use Advanced parameters in web version.
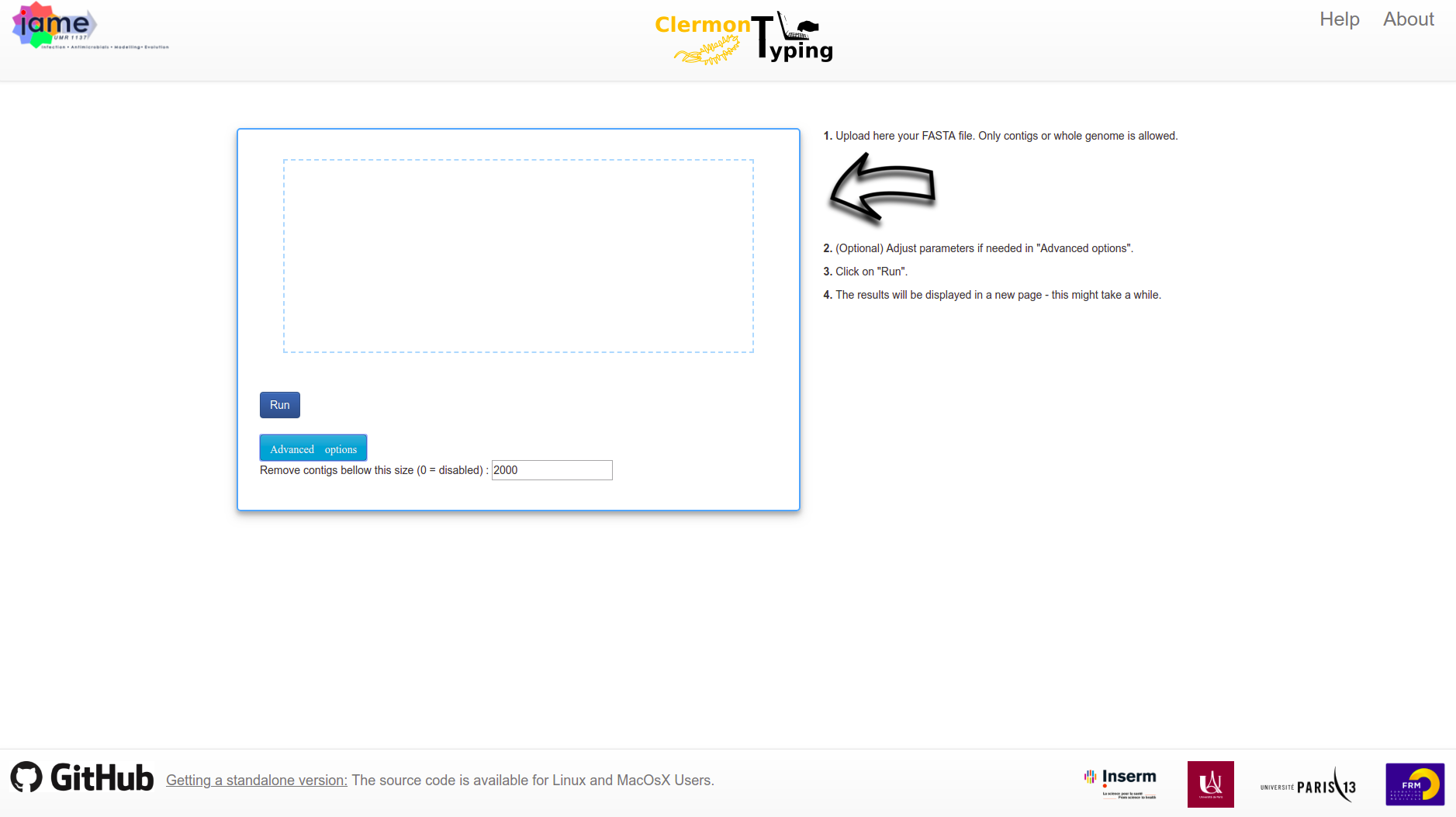
In this section you will have access to the only customizable option: the contig size cutoff. It will remove all contigs bellow a specific treshold.
By default the minimum contig size is set to 2000 bases. But setting to 0 will disable this option. If you completely trust your assembly you can set it to 0. Otherwise it is recommended to let the default value.
Interpret output
The result page report consist in one table, each line is a file you have uploaded. The conclusion of the clermont typing method is in the second to last column.
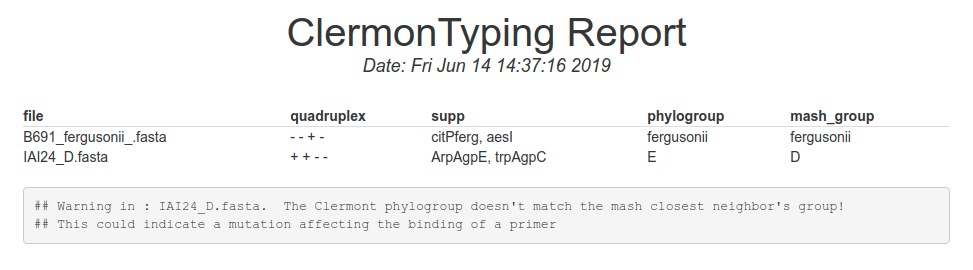
The supp column This column show the alleles specific for Escherichia albertii (chuAalbertii), Escherichia fergusonii (citPferg), then for alle specific of clades (aesI, aesII, chuIII, chuIV, chuV) and lastly for groups C and E (trpAgpC and ArpAgpE). In the exemple the IAI24 gives D or E. because we found an amplification on the ArpAgpE gene, we assigned IAI24 to the phylogroup E.
The mash group column The mash results gives an alternative method for detecting phylogroups. It is based on the whole genome sequence and is less sensible to contaminations and horizontal transferts. A star next to the mash_group results may indicate that the input sequence is a mix between multiple E. coli genomes (i.e a metagenome or a recombined genome).
What should I do if phylogroup and mash columns are different?
It may indicate that your sample is contaminated by another one (in this exemple IAI24 is suspect because the phylogroup column gives E but mash column gives D). You can increase the value of contig size cutoff in advanced parameters. Further analyses can also be done such as a recombination events detection pipeline.More informations for the output Please consider using the standalone version of clermonTyping for getting more results on the output.